SAMtools
SAM,即Sequence Alignment/Map, 测序比对结果文件格式。无论是基因组、转录组、还是表观组几乎所有流程都会产生SAM/BAM/CRAM文件作为中间步骤,然后是后续分析过程。
下载
在 官网 上下载最新版本。这里在 souceforge 上下载:

下载完后一共是三个文件
1
2
3
bcftools-1.10.2.tar.bz2
htslib-1.10.2.tar.bz2
samtools-1.10.tar.bz2
其中
bcftools-1.10.2.tar.bz2是处理BCF或VCF文件。htslib-1.10.2.tar.bz2是处理HTS的C接口,samtools-1.10.tar.bz2是我们最后需要使用的处理比对后的文件的工具。
因为samtools和bcftools都依赖htslib。但是在前两个软件包中都包含了htslib,所以不需要单独安装。
解压
1
2
3
tar -xvf bcftools-1.10.2.tar.bz2
tar -xvf htslib-1.10.2.tar.bz2
tar -xvf samtools-1.10.tar.bz2
解压后的目录结构:
1
2
3
drwxr-sr-x 8 baiyong ST_MCHRI_BIGDATA 41472 Dec 19 18:06 bcftools-1.10.2
drwxr-sr-x 7 baiyong ST_MCHRI_BIGDATA 41472 Dec 19 18:05 htslib-1.10.2
drwxr-sr-x 9 baiyong ST_MCHRI_BIGDATA 41472 Dec 7 00:46 samtools-1.10
在 安装说明有关于--with-htslib=DIR的说明:
By default, configure looks for an HTSlib source tree within or alongside the samtools source directory; if there are several likely candidates, you will have to choose one via this option.
但是目前的samtools源码中已经带了htslib。
安装
依次运行如下命令:
1
2
3
4
cd samtools-1.10
./configure --prefix=/absolute/path/to/install-location
make # 也可以加上option进行并行编译: -j 8
make install
安装完成后的结果:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
[baiyong@this-machine ~]$ samtools
Program: samtools (Tools for alignments in the SAM format)
Version: 1.10 (using htslib 1.10)
Usage: samtools <command> [options]
Commands:
-- Indexing
dict create a sequence dictionary file
faidx index/extract FASTA
fqidx index/extract FASTQ
index index alignment
-- Editing
calmd recalculate MD/NM tags and '=' bases
fixmate fix mate information
reheader replace BAM header
targetcut cut fosmid regions (for fosmid pool only)
addreplacerg adds or replaces RG tags
markdup mark duplicates
-- File operations
collate shuffle and group alignments by name
cat concatenate BAMs
merge merge sorted alignments
mpileup multi-way pileup
sort sort alignment file
split splits a file by read group
quickcheck quickly check if SAM/BAM/CRAM file appears intact
fastq converts a BAM to a FASTQ
fasta converts a BAM to a FASTA
-- Statistics
bedcov read depth per BED region
coverage alignment depth and percent coverage
depth compute the depth
flagstat simple stats
idxstats BAM index stats
phase phase heterozygotes
stats generate stats (former bamcheck)
-- Viewing
flags explain BAM flags
tview text alignment viewer
view SAM<->BAM<->CRAM conversion
depad convert padded BAM to unpadded BAM
初步使用
由于项目中使用的是.cram格式的比对结果,且已经过了排序、过滤(rmdup)和建立索引的处理,所以只需要使用samtools读取.cram并进行下游分析即可。
目前看来,在进行下游处理之前必须要先将 .cram 转换回 .bam文件,使用samtools view命令。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
[baiyong@this-machine ~]$ samtools view
Usage: samtools view [options] <in.bam>|<in.sam>|<in.cram> [region ...]
Options:
-b output BAM
-C output CRAM (requires -T)
-1 use fast BAM compression (implies -b)
-u uncompressed BAM output (implies -b)
-h include header in SAM output
-H print SAM header only (no alignments)
-c print only the count of matching records
-o FILE output file name [stdout]
-U FILE output reads not selected by filters to FILE [null]
-t FILE FILE listing reference names and lengths (see long help) [null]
-X include customized index file
-L FILE only include reads overlapping this BED FILE [null]
-r STR only include reads in read group STR [null]
-R FILE only include reads with read group listed in FILE [null]
-d STR:STR
only include reads with tag STR and associated value STR [null]
-D STR:FILE
only include reads with tag STR and associated values listed in
FILE [null]
-q INT only include reads with mapping quality >= INT [0]
-l STR only include reads in library STR [null]
-m INT only include reads with number of CIGAR operations consuming
query sequence >= INT [0]
-f INT only include reads with all of the FLAGs in INT present [0]
-F INT only include reads with none of the FLAGS in INT present [0]
-G INT only EXCLUDE reads with all of the FLAGs in INT present [0]
-s FLOAT subsample reads (given INT.FRAC option value, 0.FRAC is the
fraction of templates/read pairs to keep; INT part sets seed)
-M use the multi-region iterator (increases the speed, removes
duplicates and outputs the reads as they are ordered in the file)
-x STR read tag to strip (repeatable) [null]
-B collapse the backward CIGAR operation
-? print long help, including note about region specification
-S ignored (input format is auto-detected)
--no-PG do not add a PG line
--input-fmt-option OPT[=VAL]
Specify a single input file format option in the form
of OPTION or OPTION=VALUE
-O, --output-fmt FORMAT[,OPT[=VAL]]...
Specify output format (SAM, BAM, CRAM)
--output-fmt-option OPT[=VAL]
Specify a single output file format option in the form
of OPTION or OPTION=VALUE
-T, --reference FILE
Reference sequence FASTA FILE [null]
-@, --threads INT
Number of additional threads to use [0]
--write-index
Automatically index the output files [off]
--verbosity INT
Set level of verbosity
可以在samtools view --write-index使得输出自动输出index:
1
samtools view -T ${hg38_ref} -@ 20 --write-index ${test_cram} -b -o ${output_bam}##idx##${output_bai}
其中-T参数就指定的参考基因组,这里是Hg38,--write-index 是指明是否同时生成.bam的所以文件开关,-o 指明输出文件,它有两部分组成,前面一部分${output_bam}是生成.bam文件名,后面一部分${output_bai}是生成对应的索引文件名。他们中间用##idx##隔开。
输出结果如下:
1
2
-rw-r--r-- 1 baiyong ST_MCHRI_BIGDATA 3.5M Mar 4 16:29 sorted.rmdup.realign.BQSR.bai
-rw-r--r-- 1 baiyong ST_MCHRI_BIGDATA 1.3G Mar 4 16:29 sorted.rmdup.realign.BQSR.bam
比对结果统计
1
samtools stats ${output_bam} -@ 20 > ${output_bamstat}
得到从.bam (从.cram转换而来)的统计结果,它与原来转换之前的.bam得到的统计结果一致,说明从.cram生成.bam的过程中没有发生错误。
SAMtools 直接读取cram文件
.cram文件是.bam文件进一步压缩后的格式,在直接使用samtools读取.cram文件的时候,需要显式提供原来从.bam压缩到.cram的参考基因组.fasta文件。比如上面的例子。
如果没有显式提供参考基因组的.fasta文件,samtools所依赖的htslib会读取.cram文件头的 @SQ以确定参考基因组的MD5sum(MD5:tag)和本地参考基因组文件(UR: tag)) 。
总的来说,samtools搜索参考基因组文件的顺序为1:
- 首先看命令行是否显式指定了
-T参数, - 如果1没有找到,再去从环境变量
REF_CACHE找MD5, - 如果2没有找到,在去从环境变量
REF_PATH找MD5的每个元素, - 以上都没有,就在
.cram的UR头标签找本地参考基因组。
在实际使用的时候回出现一个问题,就是由于htslib在默认的REF_PATH被指向到http://www.ebi.ac.uk/ena/cram/md5/%s ,所以直接读取.cram的时候会出现以下错误:
1
[W::find_file_url] Failed to open reference "https://www.ebi.ac.uk/ena/cram/md5/8ae2f8b5f30a6c1762de005819965ec7": Protocol not supported
为了避免每次使用samtools的时候需要指定-T参数,同时避免samtools去http://www.ebi.ac.uk/ena/cram/md5/%s 查找参考基因文件,可以修改环境变量REF_PATH和REF_CACHE,有两种方式:
参考2的官方的方法如下:
1 2 3
<samtools_src_dir>/misc/seq_cache_populate.pl -root /some_dir/cache hg38.fasta export REF_PATH=/some_dir/cache/%2s/%2s/%s:http://www.ebi.ac.uk/ena/cram/md5/%s export REF_CACHE=/some_dir/cache/%2s/%2s/%s
这种方法比较暴力,直接在环境变量
REF_PATH和REF_CACHE中指定本地参考基因组文件:1 2
export REF_PATH=hg38.fasta export REF_CACHE=hg38.fasta
推荐使用官方提供的第一种方法。第二种方法比较暴力但实测是work的。
SAMtools查看bam/cram文件
SAMtools查看bam或cram文件格式:samtools view [-h] <.bam or .cram file> [chr1:start_position-end_position], 如
1
samtools view -h in.cram | head -10
其中 -h表示查看头文件
结果如下:
1
2
3
4
5
6
7
8
@HD VN:1.5 GO:none SO:coordinate
@PG ...
.....
@PG ID:samtools.2 PN:samtools PP:GATK PrintReads VN:1.10 CL:samtools view -h in.cram
@SQ SN:chr1 LN:248956422 M5:6aef897c3d6ff0c78aff06ac189178dd UR:/path/to/hg38.fasta
@SQ SN:chr2 LN:242193529 M5:f98db672eb0993dcfdabafe2a882905c UR:/path/to/hg38.fasta
@SQ SN:chr3 LN:198295559 M5:76635a41ea913a405ded820447d067b0 UR:/path/to/hg38.fasta
......
以上头部信息可以看出.cram文件的MD5信息和参考基因组信息:M5和URtag。
查看比对结果:
1
2
3
4
5
samtools view in.cram | head -2
#或者
samtools view in.cram chr1 | less
#或者
samtools view in.cram chr1 | more
结果如下:
1
2
CL100093941L1C010R033_411045 16 chr1 10002 0 35M * 0 0 AACCCTAACCCTAACCCTAACCCTAACCCTAACCC @?@B@ED>FEHFDDFGHGEEFGHFEDEEGEB@@@? X0:i:852 BD:Z:NOLPSOOOLPOLMNKONKLMJOOLMNLPPNPOLMM XG:i:0BI:Z:OPMPTSQROQQPOPLOOONOLOOONOLOOOOPMNN XM:i:0 XO:i:0 XT:A:R MD:Z:35 NM:i:0 RG:Z:CL100093941_L01_12
CL100093941L1C006R074_221342 0 chr1 10006 0 35M * 0 0 CTAACCCTAACCCTAACCCTAACCCTAACCCTAAC ?@@BCDDEFEFEEFFFFFEFFCFBAEFEEDBAA?A X0:i:831 BD:Z:MMOMPPLOOLMNKNMKLMJNNLMNKOOMOQOOOMN XG:i:0BI:Z:NNPNONLOOMNNLOOMNNLOONOONQRPQRQPPNO XM:i:0 XO:i:0 XT:A:R MD:Z:35 NM:i:0 RG:Z:CL100093941_L01_12
解释如下3:
QNAME 比对片段的(template)的编号;read name,read的名字通常包括测序平台等信息.
eg., ILLUMINA-379DBF:1:1:3445:946#0/1
FLAG 位标识,template mapping情况的数字表示,每一个数字代表一种比对情况,这里的值是符合情况的数字相加总和;flag取值见备注(3)
eg.16
- RNAME 参考序列的编号,如果注释中对SQ-SN进行了定义,这里必须和其保持一致,另外对于没有mapping上的序列,这里是’*‘; eg.chr1
POS 比对上的位置,注意是从1开始计数,没有比对上,此处为0;
eg.36576599
MAPQ mapping的质量,,比对的质量分数,越高说明该read比对到参考基因组上的位置越唯一;
eg.42, 如果是unmapped read则MAPQ为0。
CIGAR 简要比对信息表达式(Compact Idiosyncratic Gapped Alignment Report),其以参考序列为基础,使用数字加字母表示比对结果,match/mismatch、insertion、deletion 对应字母 M、I、D。比如3S6M1P1I4M,前三个碱基被剪切去除了,然后6个比对上了,然后打开了一个缺口,有一个碱基插入,最后是4个比对上了,是按照顺序的;
eg.36M 表示36个碱基在比对时完全匹配
注:第七列到第九列是mate的信息,若是单末端测序这几列均无意义###
RNEXT 配对片段(即mate)比对上的参考序列的编号,没有另外的片段,这里是’*‘,同一个片段,用’=’;
eg.*
PNEXT 配对片段(即mate)比对到参考序列上的第一个碱基位置,若无mate,则为0;
eg.0
TLEN Template(文库插入序列)的长度,最左边得为正,最右边的为负,中间的不用定义正负,不分区段(single-segment)的比对上,或者不可用时,此处为0(ISIZE,Inferred fragment size.详见Illumina中paired end sequencing 和 mate pair sequencing,是负数,推测应该是两条read之间的间隔(待查证),若无mate则为0);
eg.0
- SEQ 序列片段的序列信息,如果不存储此类信息,此处为’*‘,注意CIGAR中M/I/S/=/X对应数字的和要等于序列长度;
eg.CGTTTCTGTGGGTGATGGGCCTGAGGGGCGTTCTCN
- QUAL 序列的质量信息,read质量的ASCII编码。,格式同FASTQ一样。
eg.PY[[YY_______QQQQbILKIGEFGKB
- 第十二列之后:Optional fields,以tab建分割。
eg.AS:i:-1 XN:i:0 XM:i:1 XO:i:0 XG:i:0 NM:i:1 MD:Z:35T0 YT:Z:UU
查看map和unmap的reads数量
- map的reads数量
1
samtools view -F 4 -c out.bam chr21
输出为21号染色体的map的reads数:228393
- unmap的reads数量
1
samtools view -f 4 -c out.bam chr21
输出为21号染色体的unmap的reads数:0 (这是在质控的时候已经过滤掉了unmap的reads)。
查看比对的统计信息
1
samtools stats in.sorted.rmdup.realign.BQSR.cram [chr21]
结果如下:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
# This file was produced by samtools stats (1.10+htslib-1.10) and can be plotted using plot-bamstats
# This file contains statistics for all reads.
# The command line was: stats in.sorted.rmdup.realign.BQSR.cram chr21
# CHK, Checksum [2]Read Names [3]Sequences [4]Qualities
# CHK, CRC32 of reads which passed filtering followed by addition (32bit overflow)
CHK aeba9178 3e075da6 8a941b5c
# Summary Numbers. Use `grep ^SN | cut -f 2-` to extract this part.
SN raw total sequences: 10000
SN filtered sequences: 0
SN sequences: 10000
SN is sorted: 1
SN 1st fragments: 10000
SN last fragments: 0
SN reads mapped: 10000
SN reads mapped and paired: 0 # paired-end technology bit set + both mates mapped
SN reads unmapped: 0
SN reads properly paired: 0 # proper-pair bit set
SN reads paired: 0 # paired-end technology bit set
SN reads duplicated: 0 # PCR or optical duplicate bit set
SN reads MQ0: 8957 # mapped and MQ=0
SN reads QC failed: 0
SN non-primary alignments: 0
SN total length: 350000 # ignores clipping
SN total first fragment length: 350000 # ignores clipping
SN total last fragment length: 0 # ignores clipping
SN bases mapped: 350000 # ignores clipping
SN bases mapped (cigar): 350000 # more accurate
SN bases trimmed: 0
SN bases duplicated: 0
SN mismatches: 2289 # from NM fields
SN error rate: 6.540000e-03 # mismatches / bases mapped (cigar)
SN average length: 35
SN average first fragment length: 35
SN average last fragment length: 0
SN maximum length: 35
SN maximum first fragment length: 35
SN maximum last fragment length: 0
SN average quality: 35.0
SN insert size average: 0.0
SN insert size standard deviation: 0.0
SN inward oriented pairs: 0
SN outward oriented pairs: 0
SN pairs with other orientation: 0
SN pairs on different chromosomes: 0
SN percentage of properly paired reads (%): 0.0
SN bases inside the target: 2147483647
SN percentage of target genome with coverage > 0 (%): 0.01
# First Fragment Qualities. Use `grep ^FFQ | cut -f 2-` to extract this part.
# Columns correspond to qualities and rows to cycles. First column is the cycle number.
根据FLAG查看比对的metrics
1
samtools flagstat out.bam > flagst.fs
输出:
1
2
3
4
5
6
7
8
9
10
11
12
13
14974650 + 0 in total (QC-passed reads + QC-failed reads)
0 + 0 secondary
0 + 0 supplementary
0 + 0 duplicates
14974650 + 0 mapped (100.00% : N/A)
0 + 0 paired in sequencing
0 + 0 read1
0 + 0 read2
0 + 0 properly paired (N/A : N/A)
0 + 0 with itself and mate mapped
0 + 0 singletons (N/A : N/A)
0 + 0 with mate mapped to a different chr
0 + 0 with mate mapped to a different chr (mapQ>=5)
使用Picard’s CollectAlignmentSummaryMetrics工具在sorted bam上计算比对的
CollectAlignmentSummaryMetrics工具的参数:
- INPUT - The sorted bam file
- OUTPUT - “bam/MCL1.DL.alignment_metrics.bam”
- REFERENCE_SEQUENCE - “references/hg38.fa”
1
2
3
4
java -jar picard/picard.jar CollectAlignmentSummaryMetrics \
INPUT=bam/in.sorted.bam \
OUTPUT=bam/in.sorted.alignment_metrics.bam \
REFERENCE_SEQUENCE=references/hg38.fa
Q. What is the mismatch rate?
A. This is the fraction of bases in mapped reads that do not match the base at that position in the reference genome. This is a combination of the number of SNPs and sequencing errors. - 0.2308 %
统计bam中每条染色体的reads数
1
samtools idxstats out.bam | sed -n '1,25'p | less
结果为
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
chr1 248956422 647843 0
chr2 242193529 633894 0
chr3 198295559 519963 0
chr4 190214555 493178 0
chr5 181538259 465777 0
chr6 170805979 436205 0
chr7 159345973 413238 0
chr8 145138636 378403 0
chr9 138394717 326492 0
chr10 133797422 360537 0
chr11 135086622 351485 0
chr12 133275309 346957 0
chr13 114364328 273061 0
chr14 107043718 229433 0
chr15 101991189 219338 0
chr16 90338345 227827 0
chr17 83257441 209342 0
chr18 80373285 211714 0
chr19 58617616 135395 0
chr20 64444167 176055 0
chr21 46709983 119344 0
chr22 50818468 99298 0
chrX 156040895 398438 0
chrY 57227415 14011 0
chrM 16569 368 0
输出结果中,第二列是参考基因组上对应染色体的长度(bp),第三列是map的reads数,第四列是unmap的reads数。
如果要输出以上统计结果到文件,可以使用命令:
1
samtools idxstats out.bam | sed -n '2,25'p | cut -f 1,3 > out.reads.counts
samtools mpileup计算基于碱基的reads count,碱基质量和比对质量
samtools mpileup 主要用于基于碱基点位找变异,生成BCF或 VCF或pileup一个或多个bam或cram文件,命令如下:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
[baiyong@cngbx-login-1-2 ~]$ samtools mpileup
Usage: samtools mpileup [options] in1.bam [in2.bam [...]]
Input options:
-6, --illumina1.3+ quality is in the Illumina-1.3+ encoding
-A, --count-orphans do not discard anomalous read pairs
-b, --bam-list FILE list of input BAM filenames, one per line
-B, --no-BAQ disable BAQ (per-Base Alignment Quality)
-C, --adjust-MQ INT adjust mapping quality; recommended:50, disable:0 [0]
-d, --max-depth INT max per-file depth; avoids excessive memory usage [8000]
-E, --redo-BAQ recalculate BAQ on the fly, ignore existing BQs
-f, --fasta-ref FILE faidx indexed reference sequence file
-G, --exclude-RG FILE exclude read groups listed in FILE
-l, --positions FILE skip unlisted positions (chr pos) or regions (BED)
-q, --min-MQ INT skip alignments with mapQ smaller than INT [0]
-Q, --min-BQ INT skip bases with baseQ/BAQ smaller than INT [13]
-r, --region REG region in which pileup is generated
-R, --ignore-RG ignore RG tags (one BAM = one sample)
--rf, --incl-flags STR|INT required flags: skip reads with mask bits unset []
--ff, --excl-flags STR|INT filter flags: skip reads with mask bits set
[UNMAP,SECONDARY,QCFAIL,DUP]
-x, --ignore-overlaps disable read-pair overlap detection
-X, --customized-index use customized index files
Output options:
-o, --output FILE write output to FILE [standard output]
-O, --output-BP output base positions on reads
-s, --output-MQ output mapping quality
--output-QNAME output read names
--output-extra STR output extra read fields and read tag values
--output-sep CHAR set the separator character for tag lists [,]
--output-empty CHAR set the no value character for tag lists [*]
--reverse-del use '#' character for deletions on the reverse strand
-a output all positions (including zero depth)
-a -a (or -aa) output absolutely all positions, including unused ref. sequences
Generic options:
--input-fmt-option OPT[=VAL]
Specify a single input file format option in the form
of OPTION or OPTION=VALUE
--reference FILE
Reference sequence FASTA FILE [null]
--verbosity INT
Set level of verbosity
Note that using "samtools mpileup" to generate BCF or VCF files is now
deprecated. To output these formats, please use "bcftools mpileup" instead.
其中几个常用的参数:
-q:最小map quality,默认值是0-Q:最小base quality,默认值是13-C:调整碱基质量,推荐50,默认值是0-f:参考基因组-r:格式如:chr1:10000-100000-s:输出map quality
samtools mpileup输出的是基于参考基因组上有比对上的每个碱基点位的比对情况,输出还包括比对点位的碱基质量和比对质量。如下:
1
samtools mpileup -q 0 -Q 0 -f hg38.fasta -r chr21 -s -x -B -C 50 in.cram > pileup
输出结果为
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
chr21 5010011 C 1 ^!. ? !
chr21 5010012 C 1 . ? !
chr21 5010013 T 1 . @ !
chr21 5010014 T 1 . @ !
chr21 5010015 G 1 . E !
chr21 5010016 G 1 . E !
chr21 5010017 C 1 . F !
chr21 5010018 C 1 . D !
chr21 5010019 T 1 . F !
chr21 5010020 C 1 . G !
chr21 5010021 C 1 . E !
chr21 5010022 T 1 . F !
chr21 5010023 A 1 . E !
chr21 5010024 A 1 . F !
chr21 5010025 A 1 . D !
chr21 5010026 G 1 . H !
输出一共7列,分别为contig的名字,参考基因组里的碱基位置,参考碱基(reference base),覆盖该位点的reads的个数,覆盖该位点的碱基(read bases) ,碱基质量(base qualities)和mapping quality,之间由tab分开。
碱基质量(base qualities)和mapping quality是 Phred编码,其质量分数是对应的 ASCII码-33得到。
计算测序深度depth
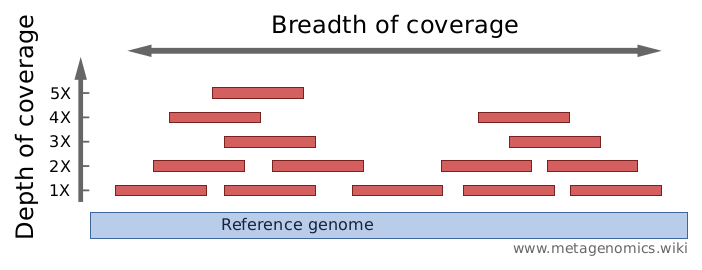
Image from 4
测序深度(Depth of coverage, mapping depth)
How strong is a genome “covered” by sequenced fragments (short reads)?
每碱基覆盖率(Per-base coverage, 每碱基测序深度)是基因组中的碱基被测序的平均次数。 基因组的覆盖深度(coverage depth of a genome)计算为与基因组比对上的所有短读的碱基总数除以该基因组的长度。 它通常表示为1X,2X,3X …(1、2、3倍覆盖率)。
覆盖范围(覆盖长度)(Breadth of coverage, covered length)
How much of a genome is “covered” by short reads? Are there regions that are not covered, even not by a single read?
覆盖范围是参考基因组被一定深度覆盖的碱基的百分比。 例如:“ 90%的基因组被1X深度覆盖;仍然70%的基因被5X深度覆盖”。
具体计算方法,参考这里.